Assignment 5
Due Date
Friday, October 5, 2012
Data Source
The data for this assignment can be found at the the Brain and Body Mass of Mammals Database web site. This web site contains a database of the brain and body masses of 1,998 mammalian species compiled from the literature.
Background
Allometry is the study of how biological processes scale with body size. The subject was pioneered by the mathematical biologist D'Arcy Thompson in the early 20th century. A common model used in allometric analysis is the so-called allometric equation, the power law relationship
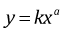
with parameters k and a. Here a is called the scaling or allometric exponent and k is called the allometric coefficient. Typically this equation is fit to data by first log transforming the equation to yield
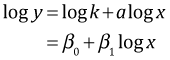
and then using ordinary linear regression (or a version of what's called Type II regression) to estimate the parameters. One of the most widely cited allometric analyses is that of Jerison (1983) who looked at the relationship between brain weight and body weight across a range of vertebrates species. Jerison argued that at higher taxonomic levels the allometric exponent is roughly the same for different taxonomic groups so that differences in the log of the allometric coefficient can be used to distinguish different organizational grades in brain architecture. In this assignment you will investigate this idea by comparing the allometric exponents of four different orders of mammals.
Questions
- Go to the Brain and Body Mass of Mammals Database and download the entire mammal database that is available there. Read the file into R.
- Tabulate the number of observations by mammalian order (Species_Order) to determine which four orders in the database have the greatest number of observations. Subset your data frame so that only those four orders of mammals are represented (discarding the rest).
- If you table a table of the species (Species_Name) counts you'll notice that some species have multiple representatives in the data base. (One species is represented 76 times!) Allometric studies have found that the magnitude of the allometric exponent decreases with taxonomic rank and approaches zero at the species level. (Thus there is no relationship between brain weight and body weight among individuals within a species.) Thus we probably should use summary statistics rather than the raw data for those species that have multiple representatives in the database.
- Obtain the mean body weight for each species in your reduced database from Question 2.
- Obtain the mean brain weight for each species in your reduced database.
- Obtain the species names and the species orders that correspond to the means you calculated in (a) and (b).
- Organize your four new variables in a single data frame.
- Carry out a formal statistical test to determine whether the allometric exponents of the brain weight to body weight relationship differ among the four mammalian orders in your data frame of means.
- If you determine that the exponents are different, determine the mammalian orders
for which the allometric exponents are significantly different and the orders for which they are not.
- Graph log mean brain weight against log mean body weight for all the observations in your database of means. Clearly differentiate the mammalian orders in your plot. Superimpose the four estimated regression equations for the four mammalian orders.
- There are at least two observations that look like they're mistakes. Identify the species corresponding to those observations.
Hints
- To simplify your life start by removing those observations in the original database that have missing values for brain mass or body mass.
- After creating your reduced database consisting of the four mammalian orders, you should redeclare Species_Name to be a factor. This will remove the ghost species names, the levels of the original Species_Name variable that no longer have any observations.
- Depending on the format you chose for downloading your original file from the mammal database, you may find that you have two orders called Cetartiodactyla. One version has a space at the end of the name. You will need to combine these two orders into a single order. There are many ways to accomplish this.
- You could use the ifelse function on the Species_Order variable checking to see if it is equal to 'Cetartiodactyla '. If TRUE assign it the correct value. If FALSE assign it its current value. The current value should be placed inside the as.character function to prevent ifelse from extracting the numeric attribute of the factor.
- Use the grep function mentioned in class yesterday to locate the offending observation and change the Species_Order of that case specifically.
- Use the sub function (see the grep help page) to replace the any space in Species_Order with nothing.
- Use the trim function from the gdata package to remove trailing spaces.
- In Question #3 there is a special R function (from a family of related functions) that we've used a few times that would be useful here.
- In Question #3 a useful generic function to use to select the taxonomic order of the unique observations that comprise your data frame of means is the following: function(x) as.character(x[1]). It selects the first value from a vector and returns its character representation.
- When you superimpose the regression lines in Question 6 be sure that you only draw each line over the range of the data that corresponds to that mammalian order. There are various ways to do this depending on the function you use to draw the lines.
- The identify function can be used to identify points in a scatter plot. It has three relevant arguments. The first argument is the x-variable in the currently active plot, the second argument is the y-variable in the currently active plot, and the third argument is the variable that contains the labels that you would like to use to label the points. Click near a point you want to identify and the corresponding label will appear at that location. To terminate the identification process on a PC right-click anywhere in the graph and choose Stop from the menu that appears. On a Mac or a PC pressing the esc key will also terminate the identification process.
Cited references
- Jerison, H. F. 1983. The evolution of the mammalian brain as an information-processing system. pp. 113–146 IN Eisenberg, J. F. & Kleiman, D. G. (Ed.), Advances in the Study of Mammalian Behavior (Spec. Publ. Amer. Soc. Mamm. 7). Pittsburgh: American Society of Mammalogists.
- Thompson, D'Arcy W. 1992. On Growth and Form (Canto ed.). Cambridge: Cambridge University Press.
Course Home Page